
The crucial role of Regulatory Affairs in the Medical Device Industry
What is Regulatory Affairs (RA) in the Medical Device (MD) Industry?
As an integral part of the healthcare world, the medical device industry must comply with a dedicated regulatory framework. These Regulatory Affairs in the Medical Device Industry aim to protect the patient and ensure health benefits.
The RA department within a medical device company is therefore responsible for :
- Identifying all regulatory texts, such as standards or guides, applicable to the MD manufactured by the company. This is called regulatory monitoring.
- Understanding the different requirements in order to respond in the most appropriate manner. Many regulatory texts are subject to interpretation. It is therefore essential to verify that the company’s understanding is in line with that of the authorities.
- Translating the requirements into the company’s internal language, adapted to each department, to ensure that they are taken into account by all employees, regardless of their position.
- Promoting and support implementation. Any change within a company is a challenge that requires resources, and therefore a real work of persuasion. It is the expert position of the RAs that allows them to convince the different teams and help them in this complex process.
- Guaranteeing the documentary management necessary for the marketing of DM manufactured by the company. Indeed, the authorities rely on documentary supports that prove or certify compliance with the applicable requirements.
Who are the different actors in the industry that interact with Regulatory Affairs?
The different tasks of the RA department imply a central position in the internal and external interactions of the company. The manufacturing of a DM is organized according to a “Supply Chain“. It includes the different departments of the company, whether technical, support or commercial, but also numerous suppliers and subcontractors. Since regulatory requirements apply to the entire manufacturing process, the RAs interact with all the players involved, particularly through regulatory audits.
The purpose of manufacturing a DM is to market it. This is only possible after having obtained a marketing authorization from the competent authorities (the Notified Bodies for Europe and the FDA (“Food and Drug Administration”) for the United States). This authorization follows an in-depth audit of the file prepared by the RAs and exchanges between the two parties.
At the same time, the role of experts implies the need to carry out a very active regulatory watch. The latest information is mainly available from the authorities and standards bodies. Nevertheless, in a proactive approach, RAs can join consortia of companies. Their aim is to exchange on the challenges they face, to define common strategies, and to promote models to the authorities, as well as within the DM community, through webinars, position papers or conventions.
What are the current challenges?
The main challenge for RAs, whatever the medical device, is to get the product on the market, and thus to put together the submission file. The documentary requirements are different depending on the authorities. Even if the content remains largely the same, the form is different, requiring a great capacity to adapt.
In addition, these files contain a lot of information about the medical device to be marketed. The large number of players involved in the supply chain makes it difficult to retrieve this data. This represents a major challenge for each person in the RA department, for which there are few solutions. Only good project management can usually provide all the information needed to form a file that meets the authorities’ expectations.
These challenges are currently even more critical in Europe following the publication of the Medical Device Regulation (MDR) 2017/745. This regulatory text requires manufacturers to resubmit all of their products in order to obtain a CE mark that complies with the new performance and safety requirements promulgated by said regulation. This process is extremely resource intensive, both in terms of human and financial resources, and has a significant impact on the viability of Medical Device companies and the product lines they sell.
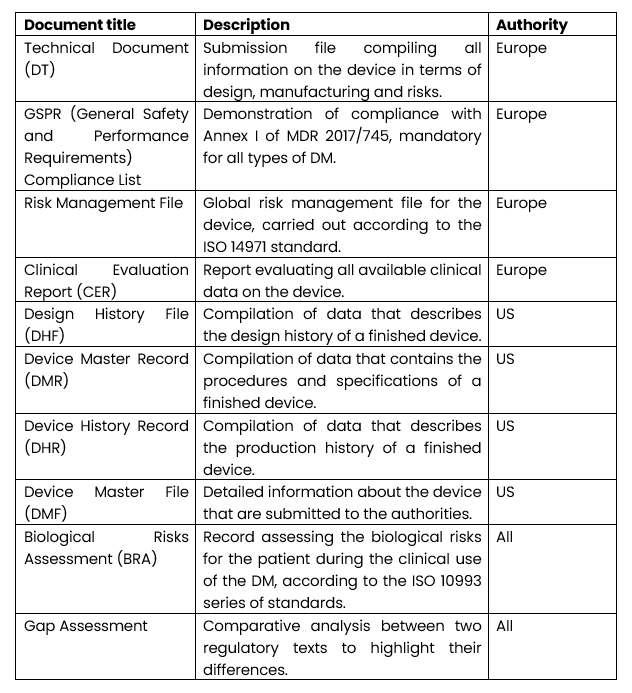
What are the main deliverables provided by RAs?
As part of the submission of a marketing file, deliverables are written to demonstrate compliance with the requirements.
The main documents managed by the RA department are presented in the table below:
How does Alispharm fit into the field of Regulatory Affairs in the Medical Device Industry?
Aware of the challenges facing industrialists today, Alispharm offers a solution: a service of experts specialized in RA.
Whether for strategic consulting or operational services, such as document drafting, Alispharm supports any structure working in the MD field in order to facilitate their success.


